CZ3253 Practical 1:
Comparative Molecular Field Analysis (CoMFA)
CoMFA Theory
The idea underlying a Comparative Molecular
Field Analysis (CoMFA) is that differences in a target property are often
related to differences in the shapes of the non-covalent fields
surrounding the tested molecules. To put the shape of a molecular field into a
QSAR table, the magnitudes of its steric (Lennard-Jones) and electrostatic (Coulombic)
fields are sampled at regular intervals throughout a defined region.
While there are many adjustable parameters in CoMFA, certainly the most
important is the relative alignment of the individual
molecules when their fields are computed. Properly aligned molecules have a
comparable conformation and a similar orientation in Cartesian space. The QSAR
is then generated by a PLS analysis of the data contained in the MSS. The value
of the resulting QSAR can be determined through the value of the
crossvalidated r2 (from now on referred to
as q2) reported by the PLS.
If acceptable, the CoMFA QSAR, re-derived in final, non-crossvalidated form, can
most easily be manipulated using various graphics techniques. Otherwise the
alignment of one or more molecules can be changed, or other parameters altered,
and the analysis repeated. Once an acceptable QSAR has been derived, prediction
of the target property value for a new molecule is particularly straightforward.
From a user's point of view, the four major
phases of a CoMFA are:
- Preparing for CoMFA
- Building Spreadsheet for CoMFA
- CoMFA PLS Runs
- Examination and Use of CoMFA Results
In this practical, we will be constructing a
QSAR for a set of ryanoid molecules. The inhibitory activity values are chicken
KDs (nM) for displacement of 7 nM [3H]ryanodine from skeletal muscle.
(W. Welch, S. Ahmad, K. Gerzon, R.A. Humerickhouse, H.R. Besch, Jr., L.
Ruest, P. Deslongchamps & J.L. Sutko, Biochemistry, 1994, 33:
6074-6085)
Preparing for CoMFA
- All the ryanoids include a tetrahydropyran
which can be used to align them. Retrieve tetrahydropyran from the Fragment
Library.
- Build/Edit >>> Get
Fragment.
- Select
TETRAHYDROPYRAN and press
OK.
- Remove all the hydrogens.
- Build/Edit >>>
Delete >>> Atom.
- Press Atom Types
in the Atom Expression dialog.
- Check the H
check box in the Atom Types dialog and press
OK.
- Press OK in the
Atom Expression dialog.
- Save the core structure as a mol2 file so
you can retrieve it later. This is a precautionary measure because the
contents of this work area will be overwritten during step 4.
- File >>> Save As.
- Enter
THP_core
in the
File
field and click on Save.
- Align all the molecules in the database
onto one of them using the molecule on the screen as the basis for the
alignment.
- File >>> Align
Database.
- The Database Align
Dialog has three fields: one for the database containing the molecules
to align, one for the template molecule, and one to indicate the
molecule area which contains the substructure common to all molecules.
Since only the default work area is currently occupied, that default
area is already entered in the appropriate text field. For subsequent
discussion, it will be assumed that this is M1.
- Type
ryanoids
in the
Database to Align
field. If there is already an entry here, you can select the whole path
by double-clicking on it and replace it. Alternatively, you can delete
the current entry and leave it empty, or even insert nonsense. Whenever
SYBYL cannot interpret the name you supply, it will launch the
Database Selection dialog.
- Type
ryanodine_1
in the
Template Molecule
field.
- Make sure the New
Database and
All Molecules
radio buttons have been checked and press
Go.
- Press OK in the
information box warning you that the contents of the molecule areas will
be overwritten during the alignment process.
- Note About Template Alignment:
Setting the Grid Orientation to
Inertial in the Database Align dialog causes the molecule upon which the
database is being aligned to be oriented along the coordinate lattice,
then frozen. This makes use of the information embedded in a CoMFA field
maximally efficient, will generally improve q2, and will make
your results reproducible by other workers. It is recommended that the
template molecule always be oriented in this way before undertaking
CoMFA.
- Watch the alignment as it proceeds: each
molecule in turn is retrieved from the database, brought into a new molecule
area and aligned on the template. Once the process is complete, you are
prompted for the name of a new database to store the 18 ryanoids in their
new orientations.
- Type
my_ryanoids
at the prompt for the name of the new database and press
OK.
- The new alignments have all been saved, so
you can now clear the screen. This will reduce the time SYBYL spends
refreshing the screen between commands.
- Build/Edit >>> Zap
(Delete) Molecule.
- Press All and
press
OK.
Building Spreadsheet for CoMFA
- Create a spreadsheet from the new database
and save it.
- Tools >>> QSAR >>>
New Spreadsheet .
- Select Database
as the Data Source and press
OK.
- In the Database
Selection dialog select
my_ryanoids.mdb
as the database and press
Open.
The 18 molecules in the
database are read in and entered as rows in the spreadsheet.
- Read in biological data
- In the end, the purpose of CoMFA is to
relate structural information to biological data. Add the relevant
biological data to your molecular spreadsheet now in preparation for
subsequent manipulations.
- MSS: File >>> Import.
- Select Custom in
the
Format
option menu of the Import dialog.
- Press the [...]
button to its right to bring up the Custom Format dialog.
- Select Comma in
the
Delimiter
option menu and toggle both
Labels check
boxes on. Enter
*
and
NEW,
respectively, in the row and column fields. This means that when the
data file is imported its rows will match the existing rows and new
columns will be created.
- Press OK to exit
the Custom Format dialog.
- Enter
ryanoids.txt
in the
File
field.
- Press Import in
the Import dialog.
- In QSAR, the logarithms of concentrations
are used. So the values we will be using are -log(concentration*10-9)
since the concentration is expressed in nM.
- Highlight column 2
and press Autofill.
- Select
FUNCTIONAL_DATA as the new column type and press
OK.
- Type
PKD
as the name for the new column and press
OK.
- Type
9-LOG("KD") for the
functional specification and press
OK.
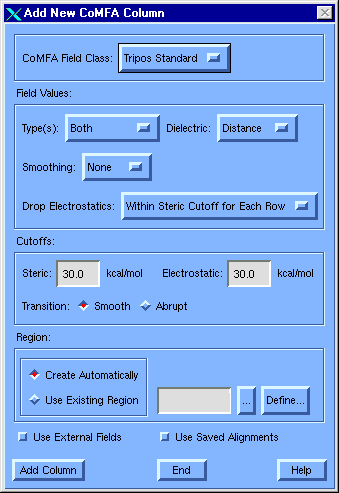
- Add CoMFA Columns
- The process of adding CoMFA fields
involves scanning all the molecules to establish an encompassing region
and computing somewhat more than 33,000 energies.
- Highlight column 3
and press Autofill.
- Select COMFA as
the new column type and press
OK.
- Select Tripos
Standard in the
CoMFA Field Class
option menu.
- Select Both as
the
Field Types.
- Make sure that the
steric and
electrostatic
cutoffs are both set to the default
30
kcal/mol, and that the Create
Automatically
radio button is on in the Region
section of the dialog.
- Press Add Column.
- Press OK to
accept
COMFA3
as the column name.
The Tripos standard CoMFA
fields are calculated for each molecule in the spreadsheet and the
number of lattice points at or above the cutoffs are displayed for each
molecule. Because thousands of actual columns of numbers are usually
produced by evaluation of a single CoMFA column, for each molecule this
column shows the number of lattice intersections located "inside" that
molecule, a very crude volume estimate.
- Question 1: What is the range of values
for the CoMFA column?
- Save the Table
- MSS: File >>> Save.
The file my_ryanoids.tbl is saved
with the CoMFA columns.
CoMFA PLS Runs
- With no rows or columns selected, any
operations are performed on the entire spreadsheet. Perform a crossvalidated
run with two objectives:
- To find out whether the CoMFA model is
predictively useful.
- If useful, decide how many components
to use for the best model. The number of components describes the degree
of complexity of the model; at some point adding more detail corresponds
to fitting the data to noise, and the predictive ability begins to
diminish.
- MSS: QSAR >>>
Partial Least Squares.
The Partial Least Squares
Analysis dialog appears.
- Column to use:
PKD, COMFA3; the only columns in the table.
- Dependent
Column: PKD; the column whose values you wish to predict from
the resulting PLS model.
- Validation:
Leave-One-Out; performs a PLS analysis with crossvalidation
where the number of groups is equal to the number of rows selected
(here all rows).
- Use SAMPLS: off;
This box is automatically checked on at start-up, but
toggle it off for now. Once you are familiar with the
crossvalidation procedure, you will probably want to use the SAMPLS
option instead, because it is much faster.
- Components:
5.
- Scaling: CoMFA
Standard; the best option when CoMFA columns are present.
- Column
Filtering: on,
2.0;
to omit from the analysis those columns (lattice points) whose
energy variance is less than 2.0 kcal/mol.
- Type
one
as the
Analysis Name.
- Press Do PLS.
A lot of details scroll in the
text window. The important summary is presented at the end: the r2
value and the optimal number of components.
Question 2: What is the crossvalidated r2? Is the model
good enough? What is the number of components for maximum r2?
- Select End
when prompted to save the analysis.
- Having the crossvalidation to confirm the
predictive ability, derive the best predictive model for use in graphics and
in numerical prediction.
- The Partial Least
Squares Analysis dialog is still on the screen. Set the options as
follows:
- Validation: No
Validation; performs a PLS analysis without any validation. This
is typically done at the end of PLS analysis.
- Components: 2.
- Scaling: CoMFA
Standard; the best option when CoMFA columns are present.
- Column
Filtering: on; to omit from the analysis those columns (lattice
points) whose energy variance is less than 2.0 kcal/mol.
- Type
two
as the Analysis Name.
- Press Do PLS.
Question 3: What is the r2?
What is the percentage of contribution to the model's information
from the electrostatic fields? What is the percentage of
contribution to the model's information from the steric fields?
- Press OK
when asked to save the analysis.
The analysis is saved in the
file two.pls.
- Press End to
close the Partial Least Squares Analysis dialog.
Examination and Use of CoMFA Results
- Investigate the CoMFA results.
- MSS: QSAR >>> View
QSAR >>> CoMFA
The View CoMFA dialog is
displayed.
- Accept the default
values for all options.
- Press Show &
Quit to exit the View CoMFA dialog.
- Read the
information in the textport window.
- The large red areas are the regions
where negative potential is favorable for the inhibitory activity, blue
areas are unfavorable. Scattered green areas are regions where bulky
substituents are desirable, yellow areas are undesirable.
- The graph in D1 represents the
predicted (Y-axis) versus Actual (X-axis) value for the dependent column
(inhibitory activity). Each point represents a compound. Compounds in
the upper right corner are the most active.
- Take a molecule that is a good candidate
for improvement, make some structural changes and predict its activity.
- Select the point
corresponding to the molecule with the highest actual value of activity.
Question 4: Which molecule has the
highest inhibitory activity? What is its actual and predicted
activities?
- Press End Select
to terminate point picking.
- Label the atoms by ID numbers.
- Press the
 icon and turn on Id Atom
Labels for the molecule in D2.
icon and turn on Id Atom
Labels for the molecule in D2.
Hydrogen 25 is near the region which
prefers negative potential (red) and near the sterically unfavorable
area (yellow). One approach to try to achieve both these objectives is
to replace the hydrogen with a fluorine.
- You can easily model this new compound.
- Build/Edit >>>
Modify >>> Atom.
- Select TYPE and
press
OK.
- Click on hydrogen 25.
- Press OK in the
Atom Expression dialog.
- Select atom type F
and press
OK.
- Calculate the charges and predict the
activity of this new compound. Note that the changes you have made to the
DehydroR_2 molecule are minor. This means that minimization and realignment
are not necessary.
- Compute >>> Charges
>>> Gasteiger-Huckel.
- Select NO
changing of formal charges.
- Build/Edit >>> Name
Molecule.
- Type
DehydroR_2F
and press
OK.
- MSS: QSAR >>>
Predict Property.
- Select
M2:DEHYDROR_2F in the molecule list and press
OK.
Question 5: What is the
predicted activity of the modified compound?
- Close the spreadsheet.
Answer to Questions
Question 1: What is the range of values for the
CoMFA column?
Answer: The values range from 154 to 236.
Question 2: What is the crossvalidated r2?
Is the model good enough? What is the number of components for maximum r2?
Answer: The crossvalidated r2 is about 0.522, so one would expect to
use such a model to account for 52% of the actual variance in activity among
additional similar ryanoids. This is good enough to at least rank the activity
of proposed new compounds rather accurately. The maximum r2 occurs at
2 components. Thus we know that the model is useful and that we should use 2
components for the final analysis.
Question 3: What is the r2? What is
the percentage of contribution to the model's information from the electrostatic
fields? What is the percentage of contribution to the model's information from
the steric fields?
Answer: Information in the text window reports that the r2 measure of
fit is 0.801. The electrostatic fields contribute 49.8% of the
model's information, while the steric fields represent the other 50.2%.
Question 4: Which molecule has the highest
inhibitory activity? What is its actual and predicted activities?
Answer: The molecule in row 5: DEHYDRO_R2 has the highest inhibitory
activity. The actual value for the inhibitory activity is 8.22, while the
predicted value is 7.85.
Question 5: What is the predicted activity of
the modified compound?
Answer: The predicted value of DehydroR_2F, the modified compound, is 8.09. The
predicted activity of the compound before modification is 7.85, as can be seen
higher in the textport window (when you picked the compound in row 5). So,
replacing the hydrogen with a fluorine is expected to have a small, positive
effect on activity.